Allen Lab for Democracy Renovation
Renovating our democratic institutions for the 21st century.
![]()
From expanding voting access to developing new strategies for antiracist change, we tackle the most pressing issues facing democracy.



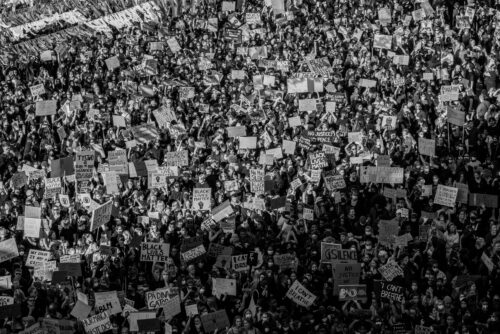

Renovating our democratic institutions for the 21st century.
If you don’t have multiracial democracy, you don’t have democracy. How can we truly achieve antiracist change?
Working to understand and foster the conditions for sustained, self-determined social and economic development among American Indian nations.
Understanding how nonviolent action can achieve democratic aims.
Reimagining our political institutions to meet the democratic challenges of today.
The Innovations in Government Program was one of the world’s premier academic entities for recognizing and promoting excellence in the public sector and fostering innovative policy solutions to the 21st-century challenges of governing. The Innovations in American Government Awards, which recognized hundreds of public-sector programs for excellence and creativity, was a principal initiative of the Program. An archive of all previous award winners remains available. Work advancing public-sector innovation continues throughout the Ash Center’s other programs and within Harvard Kennedy School. Examples of research and teaching around government innovation can be found in the Kennedy School’s programs, projects, and initiatives on cities and communities; science, technology and data; social innovation and philanthropy; public leadership and management; and much more.
The Transparency for Development Project was a novel, decade-long research initiative, housed at the Ash Center and executed in partnership with Results for Development. The Project investigated whether, why, and in what contexts local transparency and accountability interventions improve development outcomes, such as those around health and citizen participation. Specifically, T4D worked with local civil society partners in Tanzania, Indonesia, Ghana, Malawi, and Sierra Leone to implement transparency and accountability interventions along with mixed-methods evaluation, leveraging quantitative (randomized controlled trial) and qualitative (including ethnography, observations, and key informant interviews) data collection.
The project team, led by Principal Investigators Archon Fung, Jean Arkedis, Jessica Creighton, Steve Kosack, Dan Levy, and Courtney Tolmie, authored several papers, all of which are accessible as Ash Center publications as well as through Harvard’s Digital Access to Scholarship Archive. The final project results and implications are published and available here. Sierra Leone to implement transparency and accountability interventions along with mixed-methods evaluation, leveraging quantitative (randomized controlled trial) and qualitative (including ethnography, observations, and key informant interviews) data collection.